來源: 藥時(shí)空
概述
? ? 近年來��,由于國家政策的支持�����,我國的生物醫(yī)藥產(chǎn)業(yè)正在成為發(fā)展最活躍的產(chǎn)業(yè)��,醫(yī)藥研發(fā)進(jìn)展可謂日新月異��。鑒于藥監(jiān)局的監(jiān)管政策在不斷更新��,從事醫(yī)藥研發(fā)的企業(yè)和人員越來越多�����?����?紤]到從事醫(yī)藥研發(fā)的新人的增加��,有必要將新藥研發(fā)的基本流程做一個(gè)簡單梳理���。
? ? 現(xiàn)代藥物的概念除了我們傳統(tǒng)意義上的小分子化合物(如阿司匹林����,青蒿素)���,還包括多肽���、蛋白質(zhì)和抗體,(寡)核苷酸�����,小分子—抗體復(fù)合物�,還有疫苗。下面以傳統(tǒng)的小分子化合物藥為例����,就新藥(主要以1.1類創(chuàng)新藥為例)研發(fā)從無到有,到最后上市的基本流程做一個(gè)概述���。
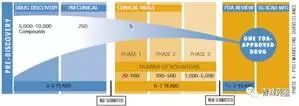
新藥的發(fā)現(xiàn)(Drug Discovery)
1.?藥物作用靶點(diǎn)(target)以及生物標(biāo)記(biomarker)的選擇與確認(rèn)
? ? 早期人們對藥物作用靶標(biāo)認(rèn)識(shí)有限����,往往只知道有效,但不知如何起效��。比如����,百年來,人們知道阿司匹林(aspirin)具有解熱�����、消炎����、止痛、抗血栓�����,甚至抗癌作用�。直到1971年,英國人John R. Vane?在《Nature》期刊發(fā)文才闡明了Aspirin作用機(jī)理為抑制前列腺素合成���,并于1982榮獲Nobel生理和醫(yī)學(xué)獎(jiǎng)?,F(xiàn)代生物醫(yī)學(xué)的研究進(jìn)展���,以及人類基因圖譜的建立��,讓人類對疾病的機(jī)理了解更加準(zhǔn)確��,為新藥開發(fā)提供了明確的方向�����、具體的靶標(biāo)��。
2.?先導(dǎo)化合物(lead compound)的確定
? ? 一旦選定了藥物作用的靶標(biāo)����,藥物化學(xué)家(medicinal chemist)首先要找到一個(gè)對該靶標(biāo)有作用的化合物���。這個(gè)化合物可以來自天然產(chǎn)物(動(dòng)物�����、植物��、海洋生物)�����;也可以是根據(jù)靶標(biāo)的空間結(jié)構(gòu)�,計(jì)算機(jī)模擬設(shè)計(jì)、合成的化合物�����;還可以根據(jù)文獻(xiàn)報(bào)道或以前其它項(xiàng)目的研究發(fā)現(xiàn)��。比如�,某一類化合物具有作用于該靶標(biāo)的藥理活性或副反應(yīng)等等���。治療勃起障礙的藥物Viagra就是由其副作用開發(fā)而成�����。目前我們常用的方法是跟蹤國外研發(fā)機(jī)構(gòu)對某一靶標(biāo)的藥物開發(fā)��,以他們的化合物作為先導(dǎo)��,希望設(shè)計(jì)出更優(yōu)秀的化合物���。
3.?構(gòu)效關(guān)系的研究與活性化合物的篩選
? ? 圍繞先導(dǎo)化合物,設(shè)計(jì)并合成大量新化合物,通過對所合成化合物活性數(shù)據(jù)與化合物結(jié)構(gòu)的構(gòu)效關(guān)系分析�����,進(jìn)一步有效的指導(dǎo)后續(xù)的化合物結(jié)構(gòu)優(yōu)化和修飾�����,以期得到活性更好的化合物���。
4.?候選藥物(candidate)的選定
? ? 通過構(gòu)效關(guān)系研究���,幾輪優(yōu)化所有篩選出來的滿足基本生物活性的最優(yōu)化合物��,一般就選作為候選藥物�����,進(jìn)入開發(fā)�。此時(shí)���,從事新藥發(fā)現(xiàn)的藥物化學(xué)家工作暫告結(jié)束�。

臨床前研究(Pre-clinic toxicology studies)
? ? 候選藥物確定后���,新藥研發(fā)就進(jìn)入開發(fā)階段(development)�,藥物開發(fā)第一階段的目標(biāo)就是完成臨床前的毒理學(xué)研究���,向藥監(jiān)部門提交“實(shí)驗(yàn)用新藥”(investigational new drug, IND?)申請。新藥開發(fā)需要多學(xué)科的協(xié)作��,比如合成工藝(process chemistry),毒理學(xué)�,藥理學(xué),藥代動(dòng)力學(xué),制劑�,等等各學(xué)科的協(xié)作,另外���,所有專業(yè)都需要分析化學(xué)的支持����。
1.?化學(xué)、制造和控制(Chemical Manufacture and Control, CMC)
? ? 新藥開發(fā)工作的第一步是原料藥合成工藝研發(fā)(Process R & D)���,這是一個(gè)不斷改進(jìn)、完善的過程�����。第一批提供的原料藥主要用于毒理研究(100—1000g),要求是越快越好�����,成本不是主要考慮因素��。因此���,只要藥化路線能夠?qū)崿F(xiàn)毒理批合成����,工藝研發(fā)部門就會(huì)采用。但隨著項(xiàng)目的推進(jìn)����,工藝部門會(huì)根據(jù)需要設(shè)計(jì)全新合成路線,開發(fā)合理生產(chǎn)工藝來滿足從I—III期臨床用藥與商業(yè)化的需求����;同理,制劑部門首先也會(huì)以最簡單的形式給藥�,完成毒理研究,然后不斷完成處方工藝研究��,開發(fā)出商業(yè)化的制劑工藝。
?2.??藥代動(dòng)力學(xué)(Pharmacokinetics, PK)
? ? 了解藥物在動(dòng)物體內(nèi)的吸收���、分布���、代謝、排泄(ADME)���,這些數(shù)據(jù)可以指導(dǎo)臨床研究以何種形式給藥(口服、吸入����、針劑),給藥頻率與劑量�。
?3.?安全性藥理(Safety Pharmacology)
? ? 證明該化合物針對特定目標(biāo)疾病具有生物活性��,同時(shí)評估藥物對療效以外的作用����,比如可能的副作用����,尤其是對心血管�、呼吸、中樞神經(jīng)系統(tǒng)的影響��。
?4.?毒理研究(Toxicology)
? ? 毒理研究種類較多�����,包括急性毒性���、亞急性毒性�����、慢性毒性���、生殖毒性、致癌性���、致突變性等��。為了加速新藥能及早驗(yàn)證是否有療效��,尤其是對一些抗癌藥����,有些耗時(shí)費(fèi)錢的毒理實(shí)驗(yàn)(如致癌性、生殖毒性)是可容許在臨床試驗(yàn)階段再進(jìn)行�。
?5.?制劑開發(fā)
? ? 制劑開發(fā)是藥物研發(fā)的一個(gè)重要環(huán)節(jié)。早期制劑研究并不需要完整的處方開發(fā)����,所有研究圍繞毒理學(xué)研究和一期臨床時(shí)方便給藥即可,目的是將候選藥物盡快推向臨床�。隨著項(xiàng)目推進(jìn),給藥方式和處方研究就越來越全面����。比如,有的藥胃腸吸收很差���,就需要開發(fā)為注射劑。有的藥對在胃酸里面會(huì)失去活性�,就需要開發(fā)為腸溶制劑。有的化合物溶解性不好�,也可以通過制劑來部分解決這個(gè)問題。
? ??前面這些內(nèi)容都統(tǒng)稱為臨床前研究�,是藥物開發(fā)的第一階段��。臨床前各個(gè)實(shí)驗(yàn)的步驟可不是嚴(yán)格按照上述這個(gè)順序展開��,而是一個(gè)相互包容���、相互協(xié)調(diào)的關(guān)系。比如���,原料藥工藝研發(fā)部門����,完成毒理批樣品合成后�����,就必須立即開展合成路線的選擇�,開發(fā)新的合成工藝,提供足夠量的原料藥以滿足制劑部門制劑研究用原料藥和9-12個(gè)月后開展I期臨床用藥的需求����。
? ? ? 因此,項(xiàng)目推進(jìn)是否順利����,就看各專業(yè)間的協(xié)調(diào)與配合是否密切了�。

臨床研究(Clinical studies)
? ? 當(dāng)一個(gè)化合物通過了臨床前試驗(yàn)后��,需要向藥監(jiān)部門(C)FDA提交新藥臨床研究申請(IND)���,以便可以將該化合物應(yīng)用于人體試驗(yàn)��。新藥臨床研究申請需要提供先前試驗(yàn)的材料�;以及計(jì)劃將在什么地方���,由誰以及如何進(jìn)行臨床試驗(yàn)的說明�����;新化合物的結(jié)構(gòu)��;給藥方式����;動(dòng)物試驗(yàn)中發(fā)現(xiàn)的所有毒性情況�;該化合物的制造生產(chǎn)情況����。所有臨床方案必須經(jīng)過倫理審評委員會(huì)(Institutional Review Board����,IRB)的審查和通過����,每年還必須向FDA和IRB匯報(bào)一次臨床試驗(yàn)的進(jìn)程和結(jié)果。在美國����,如果在提交申請后30天內(nèi)FDA沒有駁回申請��,那么該新藥臨床研究申請即被視為有效����,可以進(jìn)行人體試驗(yàn)。在中國則需要獲得CFDA正式批準(zhǔn)����,方可進(jìn)入臨床。
Ⅰ期臨床試驗(yàn)
? ? 在新藥開發(fā)過程中�,將新藥第一次用于人體以研究新藥的性質(zhì)的試驗(yàn),稱之為Ⅰ期臨床試驗(yàn)�。這一階段的臨床試驗(yàn)一般需要征集20-100名正常和健康的志愿者(對腫瘤藥物而言通常為腫瘤病人�����,但人數(shù)更少)���,在嚴(yán)格控制的條件下,給不同劑量(隨著對新藥的安全性了解的增加���,給藥的劑量也逐漸提高����,并可以多劑量給藥)的藥物試驗(yàn)于健康志愿者�����,住院以進(jìn)行24小時(shí)的密切監(jiān)護(hù)����,仔細(xì)監(jiān)測藥物的血液濃度、排泄性質(zhì)和任何有益反應(yīng)或不良作用�����,以評價(jià)藥物在人體內(nèi)的性質(zhì)��。同時(shí)也要通過這一階段的臨床試驗(yàn)獲得其吸收�����、分布��、代謝和排泄以及藥效持續(xù)時(shí)間的數(shù)據(jù)和資料���;以及藥物最高和最低劑量的信息��,以便確定將來在病人身上使用的合適劑量���。可見,Ⅰ期臨床試驗(yàn)是初步的臨床藥理學(xué)及人體安全性評價(jià)試驗(yàn)��,目的在于觀測人體對新藥的耐受程度和藥代動(dòng)力學(xué)����,為制定給藥方案和安全劑量提供依據(jù)。?
Ⅱ期臨床試驗(yàn)
? ? 為了證實(shí)藥品的治療作用的�,就必須在真正的病人身上進(jìn)行臨床研究,即Ⅱ期臨床試驗(yàn)��。Ⅱ期的臨床試驗(yàn)通常需要征集100-500名相關(guān)病人進(jìn)行試驗(yàn)���。其主要目的是獲得藥物治療有效性資料�。
? ? 將試驗(yàn)新藥給一定數(shù)量的病人志愿者����,評價(jià)藥物的藥代動(dòng)力學(xué)和排泄情況��。這是因?yàn)樗幬镌诨疾顟B(tài)的人體內(nèi)的作用方式與健康志愿者是不同的����,對那些影響腸���、胃、肝���、和腎的藥物尤其如此���。Ⅱ期臨床試驗(yàn)一般通過隨機(jī)盲法對照試驗(yàn)(根據(jù)具體目的也可以采取其他設(shè)計(jì)形式),對新藥的有效性和安全性作出初步評價(jià)���,并為設(shè)計(jì)Ⅲ期臨床試驗(yàn)和確定給藥劑量方案提供依據(jù)��。
Ⅲ期臨床試驗(yàn)
? ? 當(dāng)一個(gè)新藥推進(jìn)到三期臨床���,原料藥和制劑工藝研究也推進(jìn)到了相應(yīng)的階段。三期臨床用藥以商業(yè)化生產(chǎn)工藝提供臨床用藥���。一般來講�����,商業(yè)化生產(chǎn)的原料藥生產(chǎn)工藝應(yīng)該考慮以下因素:產(chǎn)品質(zhì)量����,生產(chǎn)安全性,生產(chǎn)成本����,環(huán)境影響,生產(chǎn)的穩(wěn)定性和可持續(xù)性�。
? ? Ⅲ期的臨床試驗(yàn)通常需?1000-5000名臨床和住院病人,在醫(yī)生的嚴(yán)格監(jiān)控下�,進(jìn)一步獲得該藥物的有效性資料和鑒定副作用,以及與其他藥物的相互作用關(guān)系�����。該階段試驗(yàn)一般將對試驗(yàn)藥物和安慰劑(不含活性物質(zhì))或已上市藥品的有關(guān)參數(shù)進(jìn)行對照和雙盲法試驗(yàn)(醫(yī)生和病人都不知道自己吃的是新藥����、老藥或安慰劑),在更大范圍的病人志愿者身上���,進(jìn)行擴(kuò)大的多中心臨床試驗(yàn)����。最后,根據(jù)嚴(yán)格統(tǒng)計(jì)學(xué)數(shù)據(jù)分析�����,進(jìn)一步評價(jià)藥物的有效性和耐受性(或安全性)��,決定新藥是否優(yōu)于(superior)或不差于(not inferior)市場現(xiàn)有的“老藥”�����。Ⅲ期臨床試驗(yàn)是治療作用的確證階段���,也是為藥品注冊申請獲得批準(zhǔn)提供依據(jù)的關(guān)鍵階段,該階段是臨床研究項(xiàng)目的最繁忙和任務(wù)最集中的部分���,無疑是整個(gè)臨床試驗(yàn)中最重要的一步��。三期臨床研究往往持續(xù)好幾年����。
? ? 除了對成年病人研究外��,還要特別研究藥物對老年病人,有時(shí)還要包括兒童的安全性��。一般來講���,老年病人和危重病人所要求的劑量要低一些�,因?yàn)樗麄兊纳眢w不能有效地清除藥物����,使得他們對不良反應(yīng)的耐受性更差,所以應(yīng)當(dāng)進(jìn)行特別的研究來確定劑量����。而兒童人群具有突變敏感性、遲發(fā)毒性和不同的藥物代謝動(dòng)力學(xué)性質(zhì)等特點(diǎn)�����,因此在決定藥物應(yīng)用于兒童人群時(shí)���,權(quán)衡療效和藥物不良反應(yīng)應(yīng)當(dāng)是一個(gè)需要特別關(guān)注的問題�����。在國外�,兒童參加的臨床試驗(yàn)一般放在成人試驗(yàn)的Ⅲ期臨床后才開始。如果一種疾病主要發(fā)生在兒童�,并且很嚴(yán)重又沒有其他治療方法,美國食品與藥品管理局允許Ⅰ期臨床試驗(yàn)真接從兒童開始�,即在不存在成人數(shù)據(jù)參照的情況下,允許從兒童開始藥理評價(jià)����。我國對此尚無明確規(guī)定。
? ? 上述任何一步反饋得到的結(jié)果不好�,都有可能讓一個(gè)候選藥物胎死腹中����。最悲慘的結(jié)果可能是這個(gè)項(xiàng)目就直接被取消了�。2007年,Merck有四個(gè)三期臨床藥物失敗�����。
? ? 能夠通過全部3期臨床評價(jià)而上市的新藥越來越少���,部分原因是開發(fā)出比市場上現(xiàn)有藥物綜合評價(jià)更好的新藥越來越難。而一個(gè)藥物從源頭研發(fā)到3期臨床是一個(gè)耗資巨大的過程���。公開數(shù)據(jù)表明�,平均下來一個(gè)新藥要花費(fèi)約為十億美金(1 billion US dollars)。正是因?yàn)樗幬镅邪l(fā)的耗資巨大�����,大公司花不起那么多錢同時(shí)展開多個(gè)項(xiàng)目研究�,小公司又沒有那么多的財(cái)力完成藥物研發(fā)的全部流程?�,F(xiàn)在的藥物研發(fā)的一個(gè)趨勢是�����,小公司反而能夠更好的找準(zhǔn)市場上的空缺���,開發(fā)出在臨床前研究階段具有良好表現(xiàn)的候選藥物��。這時(shí)大公司通過并購小公司或者購買專利(或者使用權(quán))����,將這個(gè)項(xiàng)目買過來繼續(xù)開發(fā)���。如果是購買專利的情況��,則會(huì)根據(jù)這個(gè)項(xiàng)目最后能夠進(jìn)展到哪個(gè)階段��,完成后相應(yīng)得再支付給小公司一筆“獎(jiǎng)金”�,叫做milestone�。
新藥申請(New drug application, NDA)
? ? 完成所有三個(gè)階段的臨床試驗(yàn)并分析所有資料及數(shù)據(jù),藥物的安全性和有效性得到了證明�����,新藥持有人則可以向藥監(jiān)部門(C)FDA提交新藥申請�。新藥申請需要提供所有收集到的科學(xué)資料��。通常一份新藥申請材料可多達(dá)100000?頁���,甚至更多!按照法規(guī)�����,F(xiàn)DA應(yīng)在6個(gè)月內(nèi)審評完新藥申請�。但是由于大部分申請材料過多,而且有許多不規(guī)范,因此往往不能在這么短的時(shí)間內(nèi)完成�����。中國藥監(jiān)局也在努力改進(jìn)工作�,期望縮短審批時(shí)間����。
批準(zhǔn)上市
? ? 新藥申請一旦獲得藥監(jiān)部門批準(zhǔn),該新藥即可正式上市銷售�����,供醫(yī)生和病人選擇���。但是新藥持有人還必須定期向藥監(jiān)部門呈交有關(guān)資料��,包括該藥物的副作用情況和質(zhì)量管理記錄����。對于有些藥物藥監(jiān)部門還會(huì)要求做第四期臨床試驗(yàn)���,以觀測其長期副作用情況��。
? ? 如果能夠走到這一步���,那么暫時(shí)可以說是大功告成了。從最開始的備選化合物走到這一步的藥物寥寥無幾�。但是批準(zhǔn)上市了并不代表這個(gè)藥物就高枕無憂了。因?yàn)檫€有后面一步���。

IV期臨床研究(藥物上市后監(jiān)測)
? ? 藥物在大范圍人群應(yīng)用后,需要對其療效和不良反應(yīng)繼續(xù)進(jìn)行監(jiān)測�����。藥監(jiān)部門要求根據(jù)這一階段的監(jiān)測結(jié)果來修訂藥物使用說明書����。這一階段研究還會(huì)涉及到的一些內(nèi)容有,藥物配伍使用的研究��,藥物使用禁忌�。如果批準(zhǔn)上市的藥物在這一階段被發(fā)現(xiàn)之前研究中沒有發(fā)現(xiàn)的嚴(yán)重不良反應(yīng),比如顯著增加服藥人群心血管疾病發(fā)生率之類的�����,藥物還會(huì)被監(jiān)管部門強(qiáng)制要求加注警告說明�,甚至下架。如Merck的抗關(guān)節(jié)炎藥物Vioxx?因增加心血管疾病風(fēng)險(xiǎn)于2004?年“主動(dòng)”撤離市場��。?
? ? 總之��,新藥研發(fā)是一個(gè)高風(fēng)險(xiǎn)����,高投入�,當(dāng)然也是高回報(bào)的行業(yè)。研發(fā)周期長����,涉及多學(xué)科、多專業(yè)的密切配合與協(xié)調(diào)���。如果一個(gè)新藥研發(fā)機(jī)構(gòu)各專業(yè)職能部門配套合理���,各專業(yè)人員能夠做好各自的本職工作,又注重各專業(yè)間的配合和與藥監(jiān)局的及時(shí)溝通��,新藥研發(fā)管理并不復(fù)雜��,只要有一個(gè)負(fù)責(zé)任的項(xiàng)目管理員,根據(jù)約定的項(xiàng)目關(guān)鍵時(shí)間節(jié)點(diǎn)及時(shí)協(xié)調(diào)各個(gè)職能部門的工作��,項(xiàng)目就可按計(jì)劃有序進(jìn)行���。
返回最上面
|




